
- Details
- Thursday, 09 February 2017
A new set of machine learning algorithms developed by University of Toronto researchers allows for quicker and more reliable generation of 3D structures of protein molecules. The algorithms could potentially revolutionize the development of drug therapies for a range of diseases, from Alzheimer's to cancer.
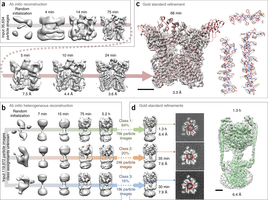
Single-particle electron cryomicroscopy (cryo-EM) is a powerful method for determining the structures of biological macromolecules. With automated microscopes, cryo-EM data can often be obtained in a few days. However, processing cryo-EM image data to reveal heterogeneity in the protein structure and to refine 3D maps to high resolution frequently becomes a severe bottleneck, requiring expert intervention, prior structural knowledge, and weeks of calculations on expensive computer clusters. Here we show that stochastic gradient descent (SGD) and branch-and-bound maximum likelihood optimization algorithms permit the major steps in cryo-EM structure determination to be performed in hours or minutes on an inexpensive desktop computer. Furthermore, SGD with Bayesian marginalization allows ab initio 3D classification, enabling automated analysis and discovery of unexpected structures without bias from a reference map. These algorithms are combined in a user-friendly computer program named cryoSPARC.
Read full article here