


Popular Articles
- Earliest molecular events of vision revealed
- Dynamics and Kinetics in Structural Biology
- XFEL Pulses Demonstrate How Plants Perceive Light
- Structural biology is solved -- now what?
- BioXFEL Postdoctoral Fellowship Award
Archived Articles
- Details
- Tuesday, 24 January 2017
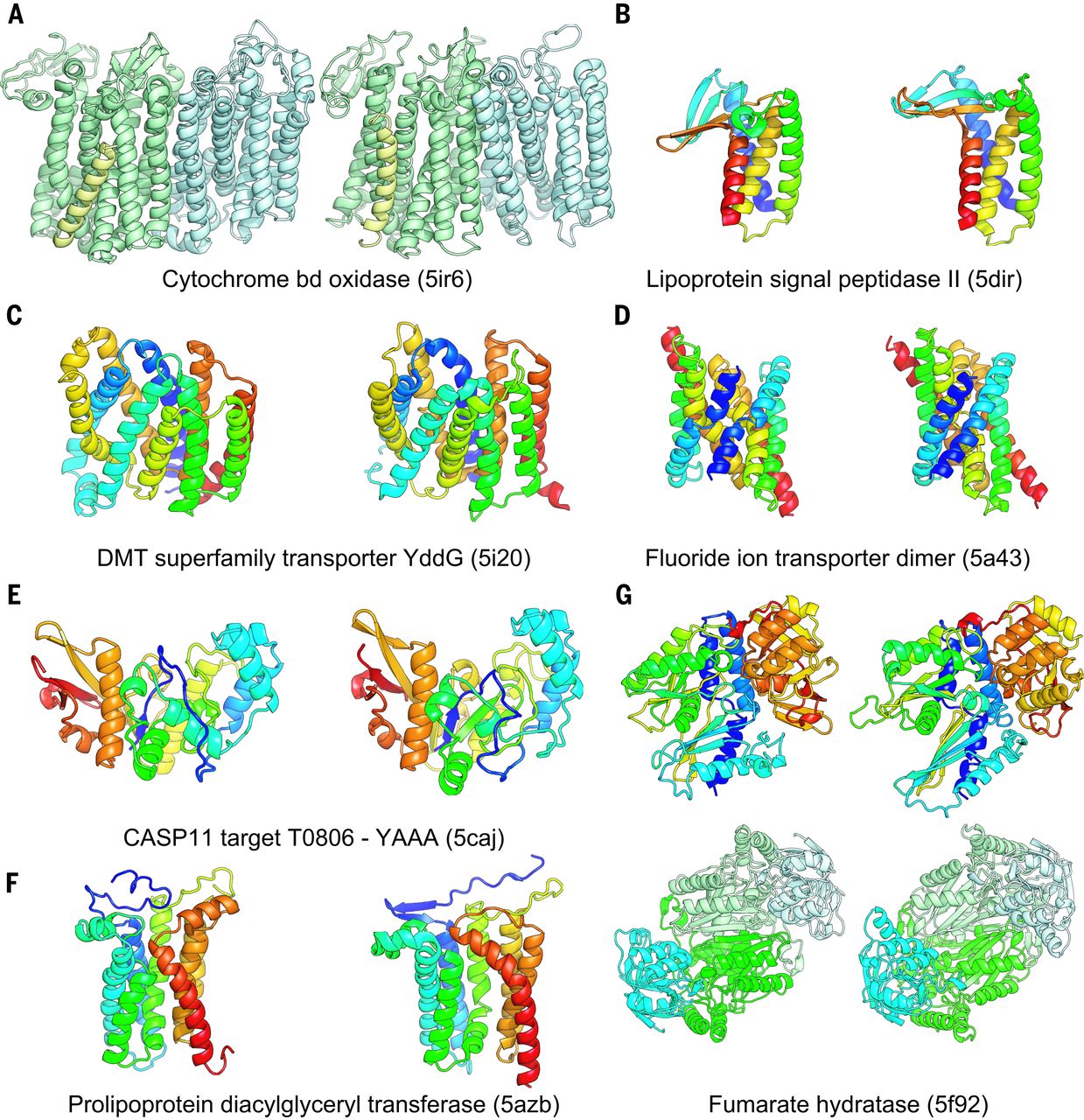
DNA sequence data collected from assorted environments has helped researchers generate 3-D models of more than 600 protein families for which the structures were previously unknown, according to a paper published in Science today (January 19). The metagenomic data enabled protein sequence comparisons across an array of species, which lent a statistical power to the predictions that would otherwise not have been possible.
“The big take-home message is that it is now possible to use computational methods to get very good models of protein structures,” said protein biochemist David Eisenberg of the University of California, Los Angeles, who was not involved in the study. “That’s a big deal because [the authors] were able to get models for many more proteins than was possible even a few years ago”
Importantly, added computational biologist Johannes Söding of the Max Planck Institute for Biophysical Chemistry in Munich, Germany, who also did not participate in the research, the “method does not need any experimental data,” such as that obtained by X-ray crystallography or nuclear magnetic resonance imaging—classical techniques for revealing a protein’s structure.
Until recently, Söding explained, biologists would predict the structures of their favorite proteins with homology modeling—“where you had a template protein that was related to the one you were interested in and from this homology you could basically copy the structure and adapt it to the new sequence.” But with the new approach, “you can now build [accurate] models even if you have no template,” he said.
Since discovering that the sequence of amino acids determines the way a protein folds, scientists have been investigating ways to calculate a protein’s structure from its sequence, said the University of Washington’s David Baker who led the new study and designed and developed protein structure–prediction software called Rosetta.
It’s known that proteins fold to their lowest energy states, said Baker, but there are often so many possible low-energy conformations, especially with large proteins, that this alone is rarely informative. Data from experimentally determined protein structures can strengthen Rosetta’s calculations, he added, “but then a few years ago we and others realized that if you had enough sequences from a large protein family, you could identify pairs of residues that were in contact in the 3-D structure based on their covariation during evolution.”
Put simply: if two amino acids interact with each other within a protein they will likely evolve together. For example, if their charges are opposite and a mutation switches the charge of one, the other will likely also switch. Comparing the sequences of a given protein from multiple species can identify such co-evolving—and thus interacting—residues.
A couple of years ago, Baker’s team used this approach to predict the structures of dozens of protein families. Since then, six of the proteins have had their structures determined experimentally and, in the new paper, side-by-side comparisons of the Rosetta predictions and the experimental results show that they are “really, really similar,” said Baker.
But what if a protein of interest doesn’t have a large array of diverse sequences available for comparison? That’s where metagenomics comes in, Baker explained.
Metagenomic data is obtained by sequencing all the DNA within a given sample—be it soil, pond water, feces, whatever—so “you get a tremendous diversity of sequences,” Baker said. “It greatly expands the number of families for which there are enough [sequence comparisons] to generate accurate models.” Indeed, using metagenomic data, the team was able to confidently predict structures for a further 614 protein families.
“It’s pretty exciting because each protein family has at least 1,000 different proteins in it, so these models cover probably over a million proteins of currently unknown structure,” Baker said.
Metagenomic data generally contain a higher proportion of prokaryotic than eukaryotic DNA sequences, said Baker. Consequently, among the remaining 4,500 or so protein families that still have no structural models, many are eukaryote-specific. “Now, what we’re trying to do is collect genome sequence data from people all over the world who are doing genome sequencing projects on eukaryotes like birds, fishes, worms, and fungi,” he said.
“The limitation is the availability of sequences,” said the University of Maryland’s John Moult who was not involved in the research. “But there are a huge number of new sequences coming out every year now, so just projecting ahead you can see that, using this same methodology, you’re going to make a big impression on the remaining families in the next five or 10 years.”
Read original article here


