


Popular Articles
- Earliest molecular events of vision revealed
- Dynamics and Kinetics in Structural Biology
- Structural biology is solved -- now what?
- XFEL Pulses Demonstrate How Plants Perceive Light
- BioXFEL Postdoctoral Fellowship Award
Archived Articles
- Details
- Wednesday, 12 July 2017
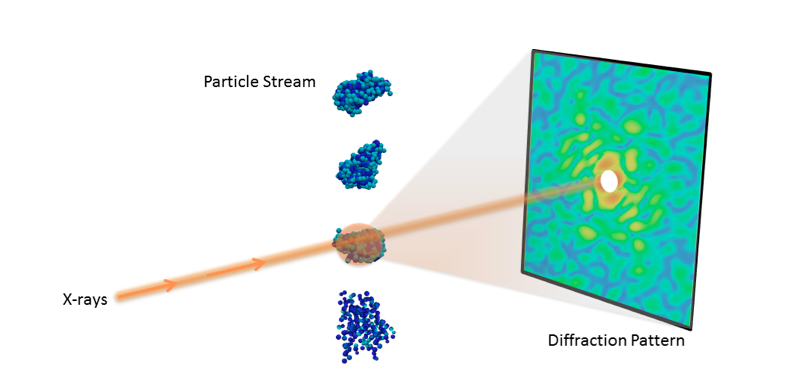
Understanding the 3D molecular structure of important nano-objects like proteins and viruses is crucial in biology and medicine. With recent advances in X-ray technology, scientists can now collect diffraction images from individual particles, ultimately allowing researchers to visualize molecules at room temperature.
However, determining 3D structure from these single-particle diffraction experiments is a significant hurdle. For instance, current data acquisition rates are very limiting, typically resulting in fewer than 10 useful snapshots per minute, limiting the amount of features that can be resolved. Additionally, the images are often highly corrupted with noise and other experimental artefacts, making it difficult to properly interpret the data.
To meet these challenges, a team of researchers from the Lawrence Berkeley National Laboratory (Berkeley Lab) has developed a new algorithmic framework called multi-tiered iterative phasing (M-TIP) that utilizes advanced mathematical techniques to determine 3D molecular structure from very sparse sets of noisy, single-particle data. This approach essentially allows researchers to extract more information from experiments with limited data. Applied mathematicians Jeffrey Donatelli and James Sethian, and physical bioscientist Peter Zwart introduced this framework by expanding on an algorithm that they originally developed to solve the reconstruction from a related X-ray scattering experiment, called fluctuation X-ray scattering. A paper describing the M-TIP framework was published June 26 in the Proceedings of the National Academy of Sciences.


