


Popular Articles
- Earliest molecular events of vision revealed
- Dynamics and Kinetics in Structural Biology
- Structural biology is solved -- now what?
- XFEL Pulses Demonstrate How Plants Perceive Light
- BioXFEL Postdoctoral Fellowship Award
Archived Articles
- Details
- Thursday, 16 March 2017
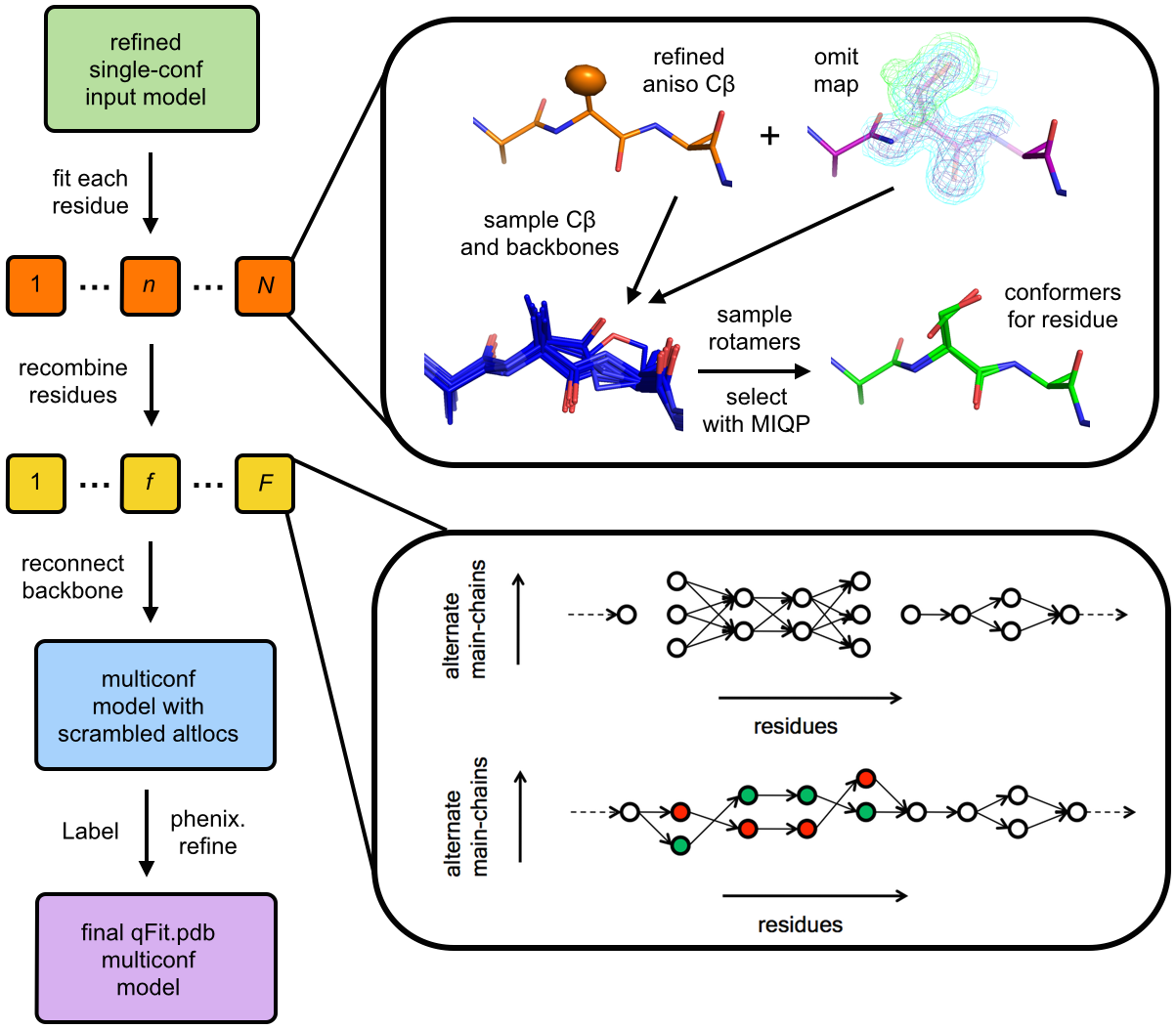
Describing the multiple conformations of proteins is important for understanding the relationship between molecular flexibility and function. However, most methods for interpreting data from X-ray crystallography focus on building a single structure of the protein, which limits the potential for biological insights. Here we introduce an improved algorithm for using crystallographic data to model these multiple conformations that addresses two previously overlooked types of protein backbone flexibility: peptide flips and glycine movements.
The method successfully models known examples of these types of multiple conformations, and also identifies new cases that were previously unrecognized but are well supported by the experimental data. For example, we discover glycine-driven peptide flips in the inhibitor-gating “flaps” of the drug target HIV protease that were not modeled in the original structures. Automatically modeling “hidden” multiple conformations of proteins using our algorithm may help drive biomedically relevant insights in structural biology pertaining to, e.g., drug discovery for HIV–1 protease and other therapeutic targets.
Proteins must move between different conformations of their native ensemble to perform their functions. Crystal structures obtained from high-resolution X-ray diffraction data reflect this heterogeneity as a spatial and temporal conformational average. Although movement between natively populated alternative conformations can be critical for characterizing molecular mechanisms, it is challenging to identify these conformations within electron density maps. Alternative side chain conformations are generally well separated into distinct rotameric conformations, but alternative backbone conformations can overlap at several atomic positions. Our model building program qFit uses mixed integer quadratic programming (MIQP) to evaluate an extremely large number of combinations of sidechain conformers and backbone fragments to locally explain the electron density. Here, we describe two major modeling enhancements to qFit: peptide flips and alternative glycine conformations. We find that peptide flips fall into four stereotypical clusters and are enriched in glycine residues at the n+1 position. The potential for insights uncovered by new peptide flips and glycine conformations is exemplified by HIV protease, where different inhibitors are associated with peptide flips in the “flap” regions adjacent to the inhibitor binding site. Our results paint a picture of peptide flips as conformational switches, often enabled by glycine flexibility, that result in dramatic local rearrangements. Our results furthermore demonstrate the power of large-scale computational analysis to provide new insights into conformational heterogeneity. Overall, improved modeling of backbone heterogeneity with high-resolution X-ray data will connect dynamics to the structure-function relationship and help drive new design strategies for inhibitors of biomedically important systems.


