


Popular Articles
- Earliest molecular events of vision revealed
- Dynamics and Kinetics in Structural Biology
- XFEL Pulses Demonstrate How Plants Perceive Light
- Structural biology is solved -- now what?
- BioXFEL Postdoctoral Fellowship Award
Archived Articles
- Details
- Monday, 29 January 2018
New research published today in Nature Methods by BioXFEL researcher Tom Grant opens up new avenues for studying the structures of particles floating in solution.
Typically, molecular structures are determined using a technique known as crystallography, where molecules are chemically induced to align next to each other in a large 3D lattice.
Bright X-rays scatter off of these molecules, greatly amplified by their strict order in the crystal, and are detected by sensitive X-ray detectors. The intensity information on these images is then used to reconstruct the 3D electron density function of the particle that can be used to determine atomic structure of the molecule. However, most particles (>75%) do not readily form ordered crystals that diffract well, leaving many molecules difficult to visualize in 3D. Additionally, biological molecules are well known to exhibit dynamic motions important for function that become constrained by the crystal lattice, resulting in the loss of important biological information.
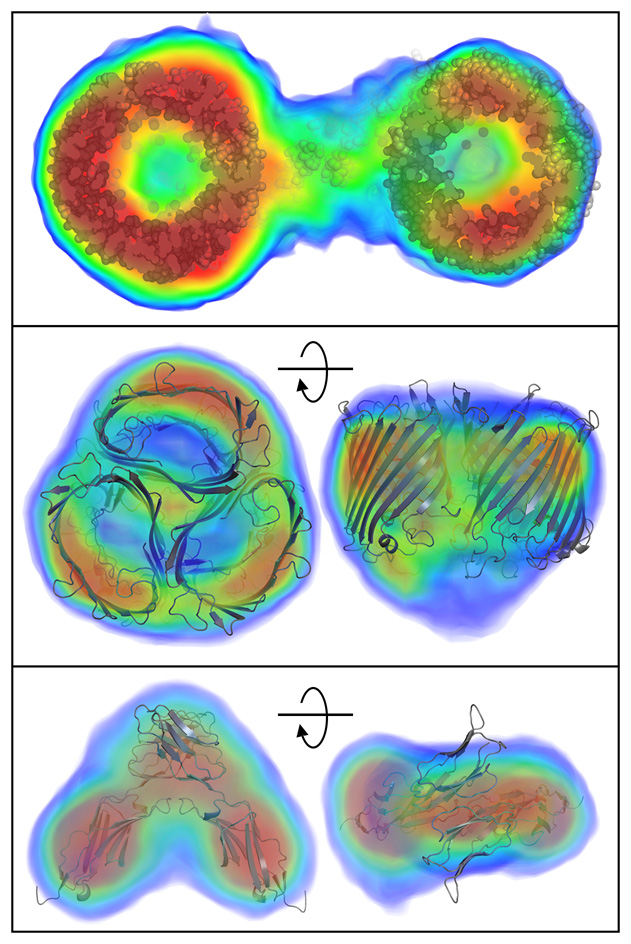
One alternative to crystallography, particularly popular for studying dynamics, is solution scattering. As the name suggests, solution scattering involves scattering the X-rays off of molecules floating in solution rather than arranged in a crystal. This allows the molecules to move dynamically in their natural states, enabling the visualization of large scale conformational dynamics important for biological function. However, as a result of the molecules tumbling in solution, the molecules scatter the X-rays in all orientations resulting in a significant loss of information as the 3D scattered intensities become 1D averaged intensities. Algorithms for extracting the 3D information from the 1D experimental data that have been developed over the last several decades have enabled the reconstruction of low-resolution molecular envelopes, outlines of the particle shapes.
The research published today in Nature Methods by BioXFEL researcher Tom Grant discusses a new algorithm that enables the reconstruction of the 3D electron density function of a molecule from the 1D solution scattering data. For the first time, this enables researchers to “see inside” these molecules floating in solution to understand the internal density variations, rather than seeing only the envelope of the particle shape. Like upgrading to a color TV from black and white, or seeing a person’s facial features rather than just a silhouette, this added information will enable researchers to better understand molecular structure in solution, in particular visualizing large scale conformational dynamics.
Grant’s method achieves this by expanding upon a well-known mathematical technique in the field of imaging known as “iterative phase retrieval”. Iterative phase retrieval, used in diverse fields such as coherent diffractive imaging, astronomy, tomography, and others, enables the reconstruction of images using only 3D intensity information (i.e. amplitudes) while missing the other important 3D information known as phase. Grant’s method, called “iterative structure factor retrieval”, expands this by reconstructing not only the 3D phases, but also the 3D amplitudes knowing only the 1D averaged intensities from the solution scattering experiment. This is the first demonstration of the ability to reconstruct 3D objects from 1D data and will likely have a large impact in these related imaging fields.
The research, published today in Nature Methods, can be found here: https://www.nature.com/articles/nmeth.4581


